R10: Investigation of Sequence–Structure Relationships in Interphase Chromatin
DNA is the largest biopolymer in nature with a length of up to 10 cm (human chromosome 1). It is bundled into a complex and highly organized structure—chromatin—to ensure efficient packing into the nucleus, which has major consequences for gene function and regulation. However, our understanding of DNA packaging and the regulatory mechanisms that take effect over long distances (up to several megabase pairs) is limited. One of the reasons for this is that modelling DNA is particularly difficult owing to its size and complexity. In biophysics, computational tools such as molecular dynamics provide structural insights only on the small-scale atomistic level. In high-throughput genomics, chromosome conformation-capturing methods merely offer coarse-grained structural information that is limited to pairwise contact probabilities. To deepen our understanding of DNA 3D structure, we plan to integrate results from bottom-up and top-down approaches. We will generate large amounts of sequencing data to produce chromatin-interaction matrices in various cell types during the developmental stage, as well as from patients with chromosomal aberrations known to cause neurodevelopmental disorders. Here, we will also consider sexual dimorphism. Experimentally, we aim at leveraging the strengths of long-read sequencing for chromatin conformation capturing to resolve 3D structures, complex rearrangements and structural variants. Subsequently, we will use these experimental data to construct chromatin models with predictive power based on concepts from molecular simulations of coarse-grained polymers. In parallel, we will use targeted simulations to identify additional parameters (e.g., epigenetic factors) that influence chromatin folding. Taking a 12-year perspective, we hope to understand how microscopic modifications (such as small mutations, genomic variants and epigenetic changes differing from cell to cell) elicit significant global macroscopic structural changes, and how these can cause human disease. To this end, we aim to establish sequencing and modelling down to the single-cell level.
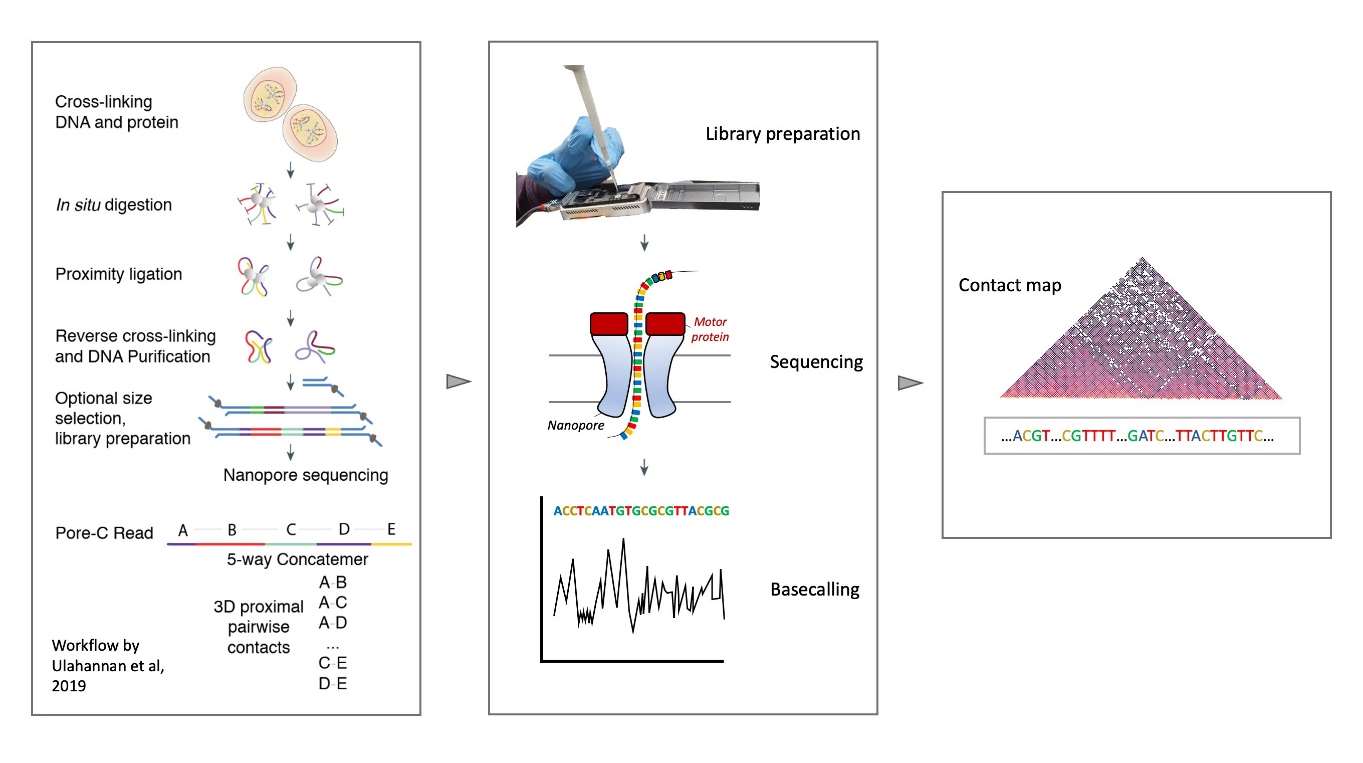
Schematic representation of the Pore-C technology. The protocol is similar to the Hi-C protocol: DNA and histones are cross-linked such that the spatial proximity of interacting loci is maintained. Restriction digestion followed by proximity ligation assembles cross-linked fragments from multiple interacting loci. These fragments are sequenced using ONT technology. By mapping each individual alignment to a single restriction fragment, a multi-path contact matrix is created, which can be decomposed into pairwise contacts.